High speed volumetric imaging with SCAPE microscopy: Extensions and applications
- Abstract number
- 389
- Event
- European Microscopy Congress 2020 Invited Speakers
- DOI
- 10.22443/rms.emc2020.389
- Corresponding Email
- [email protected]
- Session
- LST.10 - Lightsheet illumination/detection strategies to yield higher speed, higher resolution and higher throughput in Bioimaging
- Authors
- Dr Elizabeth Hillman (1)
- Affiliations
-
1. Columbia University
- Keywords
Calcium imaging, C. elegans, Drosophila, SCAPE micorscopy, Single Objective Light sheet microscopy, Zebrafish
- Abstract text
Summary
We present recent improvements to swept confocally aligned planar excitation (SCAPE) microscopy that have dramatically improved resolution, signal to noise, penetration depth and achieved volumetric imaging speeds exceeding 300 volumes per second (VPS) in living samples [1-2]. In addition to these improvements, we will also describe extensions of the technique and new applications for high-speed imaging of cellular function on the level of whole, freely moving organisms, and high-throughput imaging of large fixed, cleared and expanded tissues.
Introduction
As fluorescent indicators continue to improve and a wider range of animal models and 3D culture systems are established, there is a growing need for microscopes that can image large volumes of these diverse samples at high speeds with minimal photodamage. Swept confocally aligned planar excitation microscopy (SCAPE) meets many of these needs by leveraging the power of single objective light sheet microscopy with confocal-type scanning that enables ultra high-speed imaging of a wide range of samples. Since our first demonstration of SCAPE in 2015, we have made significant improvements to all aspects of the technique, including resolution, imaging speed, field of view and penetration depth into challenging living samples such as the larval Drosophila and intact, living mouse brain. We have also further simplified the microscope’s design to make it simpler to build, align and maintain and are assisting many groups to build their own systems.
Methods
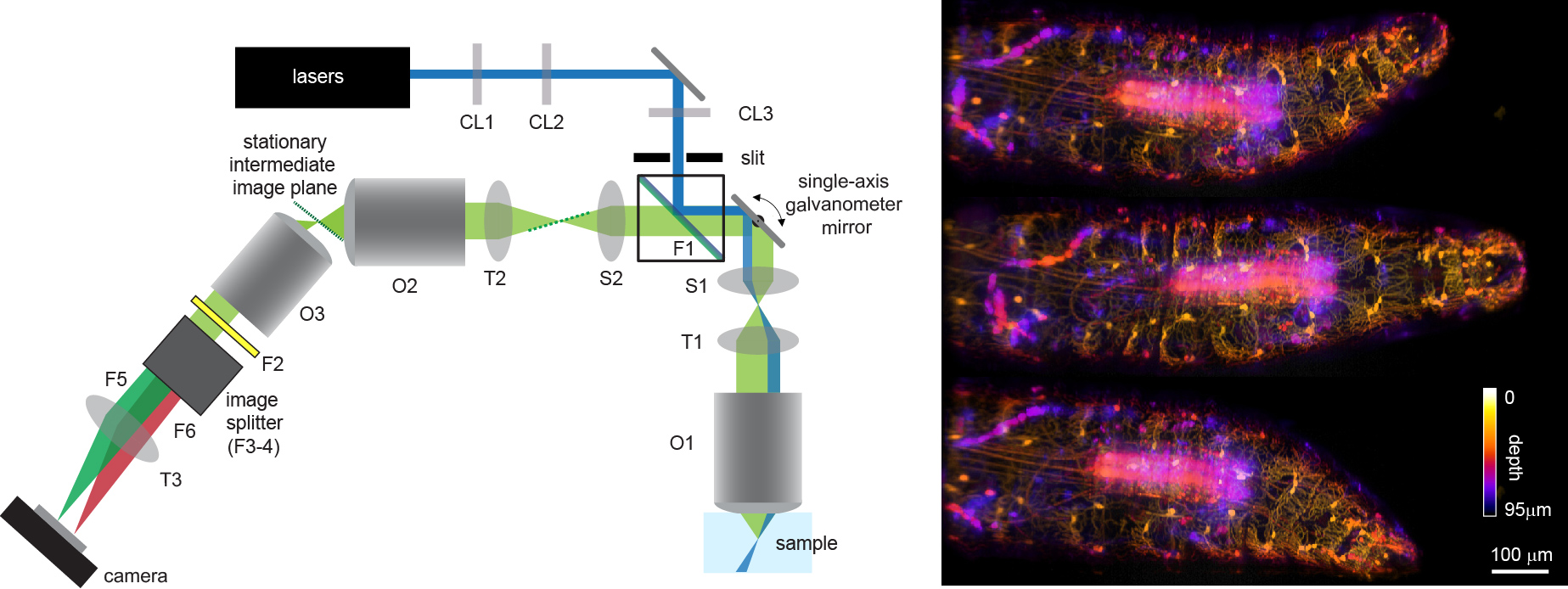
As shown in Figure 1, SCAPE microscopy works by illuminating the sample with an oblique sheet of laser light though a high NA objective lens in a standard upright or inverted microscope geometry. Fluorescence excited by this light sheet is detected back through the same objective and relayed into a second objective lens which creates an intermediate image of the oblique light sheet. A third objective lens is then oriented to be focused on this oblique intermediate image plane, generating a focused image of the sheet on a fast camera. A galvanometer mirror positioned between objectives 1 and 2, and imaged onto the back focal planes of both, causes the oblique light sheet at the sample to move from side to side along x, sampling an oblique 3D volume as a sequence of y-z’ planes. This galvanometer mirror also descans returning fluorescence light, causing the oblique image of the sheet at the intermediate image plane to stay stationary, and thus the camera to maintain its focus on the scanning oblique sheet at the sample. This arrangement generates an imaging geometry similar to standard orthogonal light sheet systems, but does not require special mounting of the sample, movement of the sample, or repositioning of the objective lens to image a 3D volume within the sample. Because the only moving part of this system is a galvanometer mirror, which only needs to move at the volume rate of the system (e.g. 10 Hz for 10 VPS) it is possible to achieve imaging speeds of over 300 VPS in a wide range of intact samples. Dual color imaging is achieved by positioning an image splitter in front of the camera, and since the number of rows on the camera corresponds to the number of depths within the sample (e.g. 200) the camera can be read out at very high speeds (> 1KHz frame rates). SCAPE shares the light efficiency and low phototoxicity benefits of conventional light sheet microscopy, making it possible to image sensitive, living samples at very high speeds for long durations with minimal photodamage.
Figure 1. Left: SCAPE schematic. CL = cylindrical lenses, (CL1 = Powell lens), O = Objective lenses, T = tube lenses, S = scan lenses (S1 is a Plossl), F = filters, F1 can be a dichroic or a small mirror. Blue at sample denotes water-immersion. Right: 3 depth-resolved images of a Drosophila larva with GFP labeling of ventral proprioceptive neurons acquired at 10 volumes per second as the larva crawled and explored.
Results
We have applied SCAPE to imaging a wide range of living samples including C. Elegans, larval zebrafish brain and heart, adult and larval fruit flies (Figure 1, Right), organoids and the living mouse brain and olfactory epithelium. In these samples we can capture both structure (from targeted fluorescent proteins) and function (e.g. from genetically encoded calcium indicators such as GCaMP). We are working on new systems including 2P-SCAPE for imaging large volumes of neuronal activity deeper in the mammalian brain.
Conclusion
SCAPE microscopy offers new capabilities for high speed 3D microscopy, offering significant advantages over confocal microscopy including 3D imaging speeds, better signal to noise and reduced photodamage.
- References
[1] Bouchard, M. B., V. Voleti, C. S. Mendes, C. Lacefield, W. B. Grueber, R. S. Mann, R. M. Bruno and E. M. Hillman (2015). "Swept confocally-aligned planar excitation (SCAPE) microscopy for high speed volumetric imaging of behaving organisms." Nat Photonics 9(2): 113-119.
[2] Voleti, V., K. B. Patel, W. Li, C. Perez Campos, S. Bharadwaj, H. Yu, C. Ford, M. J. Casper, R. W. Yan, W. Liang, C. Wen, K. D. Kimura, K. L. Targoff and E. M. C. Hillman (2019). "Real-time volumetric microscopy of in vivo dynamics and large-scale samples with SCAPE 2.0." Nat Methods 16(10): 1054-1062.